Density Functional Computations on Noble Gas Atoms

Requires a Wolfram Notebook System
Interact on desktop, mobile and cloud with the free Wolfram Player or other Wolfram Language products.
Density functional theory (DFT) has now become the predominant technique in computational quantum chemistry, having displaced wavefunction-based computations for atoms, molecules and solids. The key reason is that QFT deals with a single electron density function 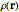 for an
for an  -electron system, rather than a complicated combination of
-electron system, rather than a complicated combination of  orbital functions
orbital functions 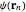 . The fundamental validity of DFT and its practical implementation by a variational principle are expressed in two theorems of Hohenberg and Kohn. For all necessary background on DFT, refer to the definitive monograph of Parr and Yang [1]. For more recent advances, see also [2].
. The fundamental validity of DFT and its practical implementation by a variational principle are expressed in two theorems of Hohenberg and Kohn. For all necessary background on DFT, refer to the definitive monograph of Parr and Yang [1]. For more recent advances, see also [2].
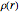
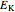
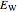
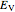
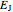
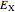
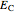
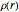

Contributed by: S. M. Blinder (June 2019)
Open content licensed under CC BY-NC-SA
Details
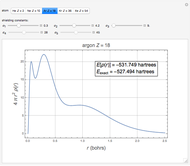
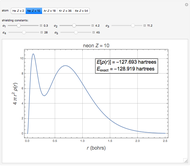
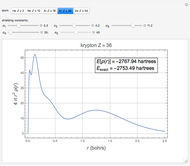
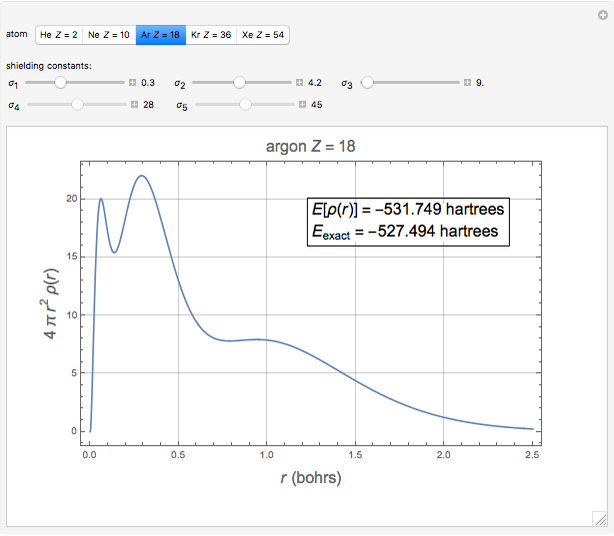
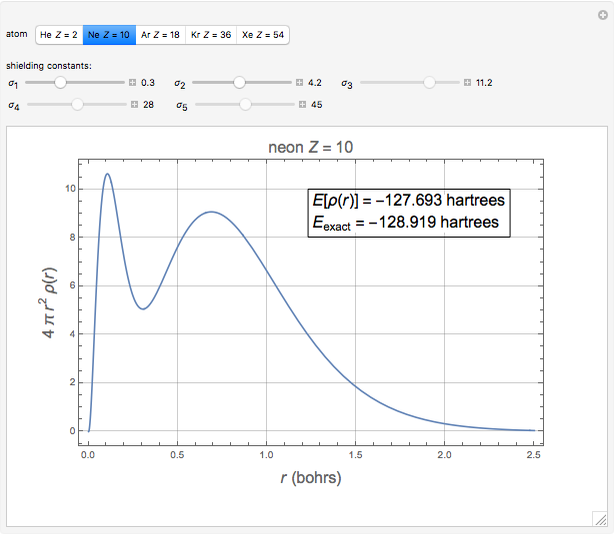
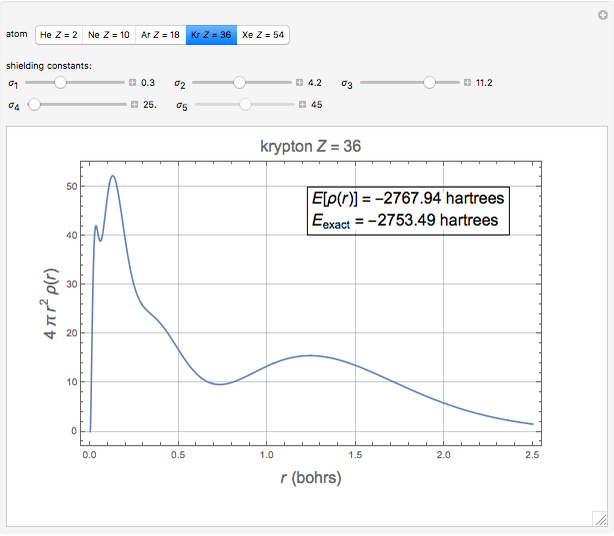
The total electron density is approximated by a sum of shells (one to five shells for He to Xe):
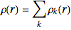 , where
, where 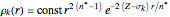 ,
,
which is suggested by Slater's rules for atomic orbitals.
The DFT functional takes the form
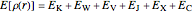 ,
,
with
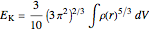 ,
,
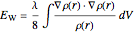 ,
, 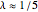 ,
,
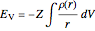 ,
,
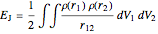 ,
,
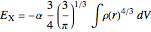 ,
, 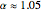 ,
,
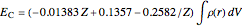 .
.
The last formula is a conjecture by the author based on computations of atomic correlation energies.
It is most convenient to carry out all the integrals numerically. The energy functional 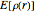 , based on the selected shielding parameters
, based on the selected shielding parameters 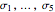 , is computed and compared with the exact (nonrelativistic) energy of the atom. By the second Hohenberg–Kohn theorem, the optimized energy for the functional form of
, is computed and compared with the exact (nonrelativistic) energy of the atom. By the second Hohenberg–Kohn theorem, the optimized energy for the functional form of 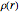 is a minimum, although short of the exact energy.
is a minimum, although short of the exact energy.
References
[1] R. G. Parr and W. Yang, Density-Functional Theory of Atoms and Molecules, New York: Oxford University Press, 1989.
[2] J. Sun, J. W. Furness and Y. Zhang, "Density Functional Theory," Mathematical Physics in Theoretical Chemistry (S. M. Blinder and J. E. House, eds.), Amsterdam: Elsevier, 2018 Chapter 4. doi:10.1016/B978-0-12-813651-5.00004-8.
[3] W.-P. Wang and R. G. Parr, "Statistical Atomic Models with Piecewise Exponentially Decaying Electron Densities," Physical Review A, 16(3), 1977 pp. 891–902. doi:10.1103/PhysRevA.16.891.
[4] S. M. Blinder. "Shell Structure of Noble Gas Atoms" from the Wolfram Demonstrations Project—A Wolfram Web Resource. demonstrations.wolfram.com/ShellStructureOfNobleGasAtoms.
Snapshots
Permanent Citation
 Shell Structure of Noble Gas Atoms
Shell Structure of Noble Gas Atoms
S. M. Blinder WKB Computations on Morse Potential
WKB Computations on Morse Potential
S. M. Blinder Hydrides as Isoelectronic Perturbations of the Neon Atom
Hydrides as Isoelectronic Perturbations of the Neon Atom
S. M. Blinder Connection between Quantum-Mechanical Hydrogen Atom and Harmonic Oscillator
Connection between Quantum-Mechanical Hydrogen Atom and Harmonic Oscillator
S. M. Blinder Generalized Unsöld Theorem for Hydrogenic Functions
Generalized Unsöld Theorem for Hydrogenic Functions
S. M. Blinder Two-Parameter Variational Functions for the Helium Isoelectronic Series
Two-Parameter Variational Functions for the Helium Isoelectronic Series
Wai-Kee Li and S. M. Blinder Temperature Variation of Heat Capacity for an Ideal Diatomic Gas
Temperature Variation of Heat Capacity for an Ideal Diatomic Gas
Hayley Petit and Siddharth Madapoosi Visualizing Atomic Orbitals
Visualizing Atomic Orbitals
Guenther Gsaller Energy Levels and Wavefunctions of Two Coupled Atomic Rings
Energy Levels and Wavefunctions of Two Coupled Atomic Rings
Vasil Saroka Low-Temperature Heat Capacity of Hydrogen Molecules
Low-Temperature Heat Capacity of Hydrogen Molecules
S. M. Blinder
-
 Polarizability of Hydrogen Atoms
Polarizability of Hydrogen Atoms
S. M. Blinder -
 Solutions of Schrödinger Equation for a Particle in a Finite Spherical Well
Solutions of Schrödinger Equation for a Particle in a Finite Spherical Well
S. M. Blinder -
 Simulated Quantum Computer Algorithm for Database Searching
Simulated Quantum Computer Algorithm for Database Searching
S. M. Blinder -
 Hanbury Brown and Twiss Interference for Bosons and Fermions
Hanbury Brown and Twiss Interference for Bosons and Fermions
S. M. Blinder -
 The Structure of Diamond
The Structure of Diamond
S. M. Blinder -
Hydrides as Isoelectronic Perturbations of the Neon Atom
S. M. Blinder -
 Zero-Energy Limit of Coulomb Wavefunctions
Zero-Energy Limit of Coulomb Wavefunctions
S. M. Blinder -
 Density Functional Computations on Noble Gas Atoms
Density Functional Computations on Noble Gas Atoms
S. M. Blinder -
 Exact Solutions of the Schrödinger Equation for the Kratzer Potential
Exact Solutions of the Schrödinger Equation for the Kratzer Potential
S. M. Blinder -
 Exact Solutions of the Schrödinger Equation for Pseudoharmonic Potential
Exact Solutions of the Schrödinger Equation for Pseudoharmonic Potential
S. M. Blinder -
Low-Temperature Heat Capacity of Hydrogen Molecules
S. M. Blinder -
 Constructing a Regular Heptadecagon (17-gon) with Ruler and Compass
Constructing a Regular Heptadecagon (17-gon) with Ruler and Compass
S. M. Blinder -
 Free Rotation of a Rigid Body: Poinsot Constructions
Free Rotation of a Rigid Body: Poinsot Constructions
S. M. Blinder -
 Venus Is Not the Earth's Closest Planetary Neighbor
Venus Is Not the Earth's Closest Planetary Neighbor
S. M. Blinder -
 Two-Dimensional Oscillator in Magnetic Field
Two-Dimensional Oscillator in Magnetic Field
S. M. Blinder -
 Charged Harmonic Oscillator in Electric Field
Charged Harmonic Oscillator in Electric Field
S. M. Blinder -
 Eigenvalues for a Pure Quartic Oscillator
Eigenvalues for a Pure Quartic Oscillator
S. M. Blinder -
 Energies of Helium Isoelectronic Series Using Perimetric Coordinates
Energies of Helium Isoelectronic Series Using Perimetric Coordinates
S. M. Blinder -
Connection between Quantum-Mechanical Hydrogen Atom and Harmonic Oscillator
S. M. Blinder -
 Dovetail Joint
Dovetail Joint
S. M. Blinder