Distribution of End-to-End Distances in Linear Substituted Polymethylenes





Requires a Wolfram Notebook System
Interact on desktop, mobile and cloud with the free Wolfram Player or other Wolfram Language products.
This Demonstration shows the dependence of the end-to-end distance distribution of a linear substituted polymethylene chain on energy barriers, temperature and chain length. The parameter values are automatically reset to force  and
and  .
.
Contributed by: A. A. Koledenkov (April 2019)
Open content licensed under CC BY-NC-SA
Snapshots
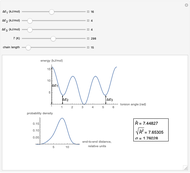
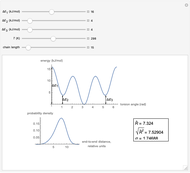
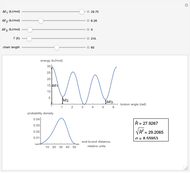
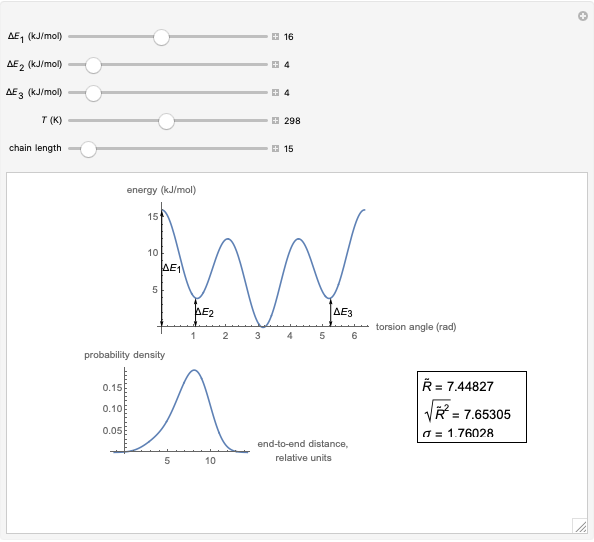
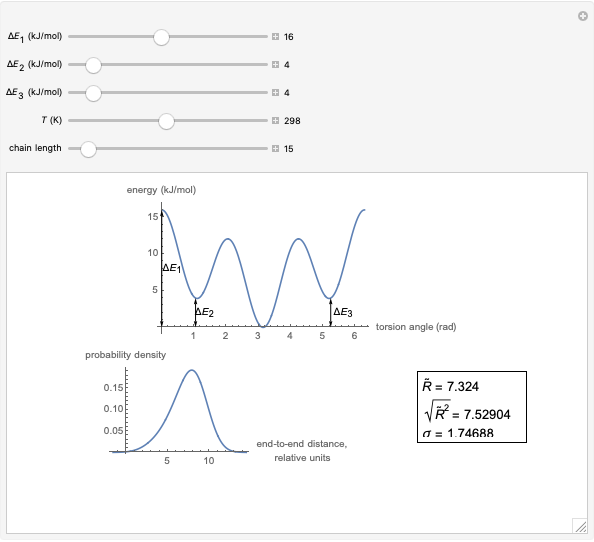
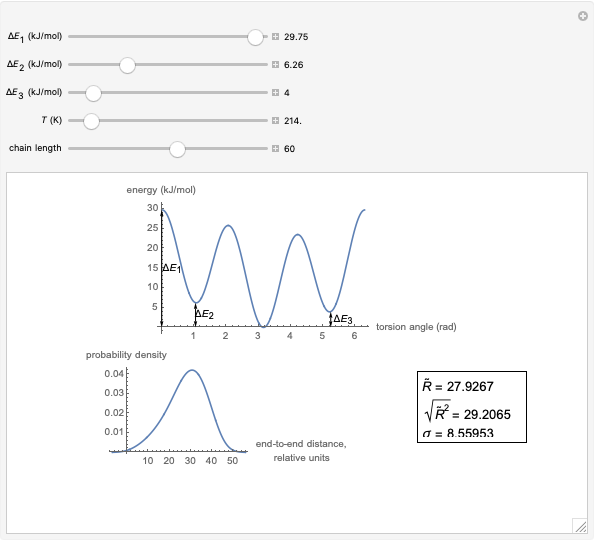
Details
The conformational energy of hindered rotation  in a classical molecular mechanics simulation in the general case is approximated by the equation [1]:
in a classical molecular mechanics simulation in the general case is approximated by the equation [1]:
 .
.
The parameters  and
and  describe a force field with torsion angle
describe a force field with torsion angle  .
.
This Demonstration uses a modified version of the potential:

where  is the energy barrier (kJ/mol) and
is the energy barrier (kJ/mol) and  and
and  are the energy difference gauche(-)/trans and gauche(+)/trans conformers, respectively (kJ/mol).
are the energy difference gauche(-)/trans and gauche(+)/trans conformers, respectively (kJ/mol).
The effect of excluded volume is neglected in the calculation.
Using the Monte Carlo method, 500 macromolecular chains are generated. For a chain of length  ,
,  torsion angle values are generated. To simplify the calculation, selection of the torsion angle is performed taking into account the statistical weight based on the cumulative distribution function
torsion angle values are generated. To simplify the calculation, selection of the torsion angle is performed taking into account the statistical weight based on the cumulative distribution function  at temperature
at temperature  , divided into 36 equal intervals with step size 10°. This avoids the calculation of the Boltzmann factor for individual chains:
, divided into 36 equal intervals with step size 10°. This avoids the calculation of the Boltzmann factor for individual chains:
 .
.
For each chain, the end-to-end distance  is calculated. The distance is expressed in relative units of carbon-carbon bond length. For visualization, a smoothed function is used to exclude statistical scatter;
is calculated. The distance is expressed in relative units of carbon-carbon bond length. For visualization, a smoothed function is used to exclude statistical scatter;  values are the result of smoothing and should be ignored.
values are the result of smoothing and should be ignored.
Based on the distribution, the mean  , root mean square
, root mean square  , end-to-end distance and standard deviation
, end-to-end distance and standard deviation  are calculated:
are calculated:
 ,
,
 ,
,
 .
.
Reference
[1] M. Bachmann, Thermodynamics and Statistical Mechanics of Macromolecular Systems, Cambridge, UK: Cambridge University Press, 2014.
Permanent Citation
 Dendrimer to Linear Polymer Transition
Dendrimer to Linear Polymer Transition
Borislav Angelov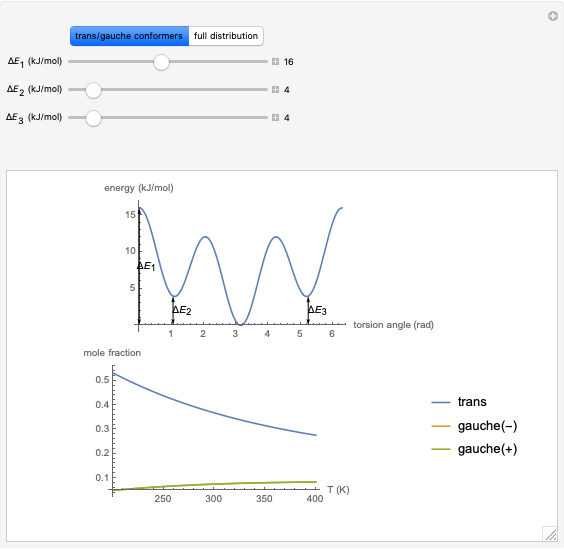 Distribution of Conformers in Rotational Isomerism
Distribution of Conformers in Rotational Isomerism
A. A. Koledenkov Substituted Pyrimidines and Purines
Substituted Pyrimidines and Purines
S. M. Blinder Electrophilic Aromatic Substitution Reactions of Benzene
Electrophilic Aromatic Substitution Reactions of Benzene
D. Meliga and S. Z. Lavagnino Thermal Distribution in an Optical Fiber with Heat Source
Thermal Distribution in an Optical Fiber with Heat Source
Brian G. Higgins and Housam Binous Spectral-Temporal Distribution of Biphotons from a Spontaneous Parametric Down Conversion (SPDC) Process
Spectral-Temporal Distribution of Biphotons from a Spontaneous Parametric Down Conversion (SPDC) Process
Ruibo Jin Rotating Beam Fatigue Tester
Rotating Beam Fatigue Tester
Erik Mahieu Normal Oscillatory Modes of Rotating Orthotropic Disks
Normal Oscillatory Modes of Rotating Orthotropic Disks
Atefeh Khoshnood and Mir Abbas Jalali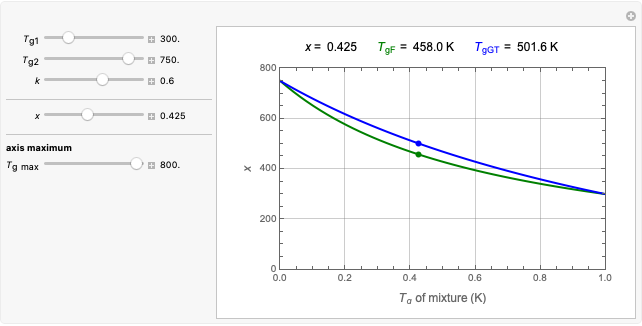 Gordon-Taylor and Fox Equations for Glass Transition Temperature
Gordon-Taylor and Fox Equations for Glass Transition Temperature
Mark D. Normand and Micha Peleg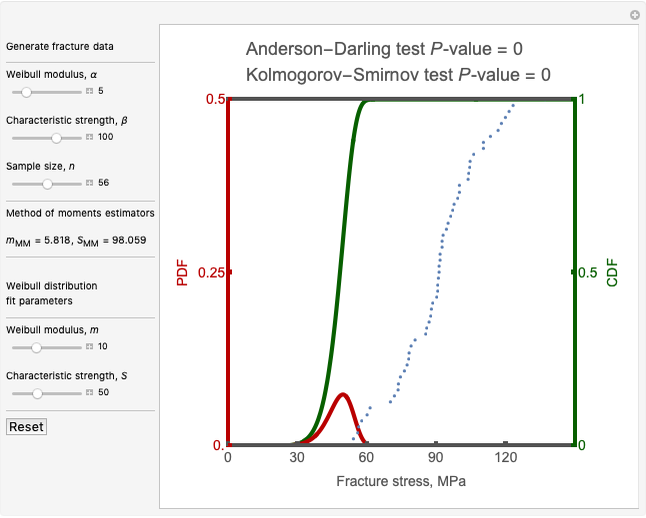 Weibull Fit to Computer Generated Fracture Data
Weibull Fit to Computer Generated Fracture Data
Ozgur Keles