Hydrogen and Lithium Orbitals Using a Hartree Eigenvalue Method

Requires a Wolfram Notebook System
Interact on desktop, mobile and cloud with the free Wolfram Player or other Wolfram Language products.
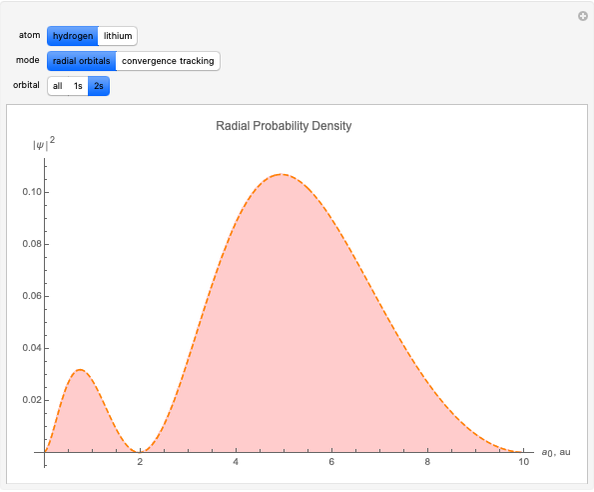
This Demonstration shows how to numerically calculate radial 1s and 2s orbitals of hydrogen and lithium. A Hartree eigenvalue method is applied to a 300×300 square matrix with a step size of  au. The method uses the fact that the interaction potential between electrons can be simplified significantly for simple atoms. Numerically solving for eigenvalues and eigenfunctions of the Schrödinger equation with such a potential is carried out using an iterative process.
au. The method uses the fact that the interaction potential between electrons can be simplified significantly for simple atoms. Numerically solving for eigenvalues and eigenfunctions of the Schrödinger equation with such a potential is carried out using an iterative process.
Contributed by: Nikita M. Kostylev (March 2011)
Open content licensed under CC BY-NC-SA
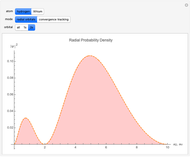
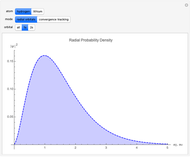
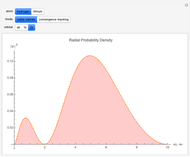
Snapshots
Details
To find the solution of a simplified Schrödinger equation for  (
( ), we can simply determine numerical eigenvalue solutions to
), we can simply determine numerical eigenvalue solutions to
 .
.
 is defined as:
is defined as:
 ,
where the potential
,
where the potential  is the interaction potential. It can be approximated [1] by
is the interaction potential. It can be approximated [1] by
 ,
,
where  is the charge, spherically symmetrical and assuming it acts as a Gaussian surface:
is the charge, spherically symmetrical and assuming it acts as a Gaussian surface:
 .
.
Establishing a transformation matrix that would represent the Hamiltonian, using three-point first- and second-order numerical derivatives for the Laplacian and solving iteratively for eigenvalues and eigenvectors generates the approximate solutions for radial density functions.
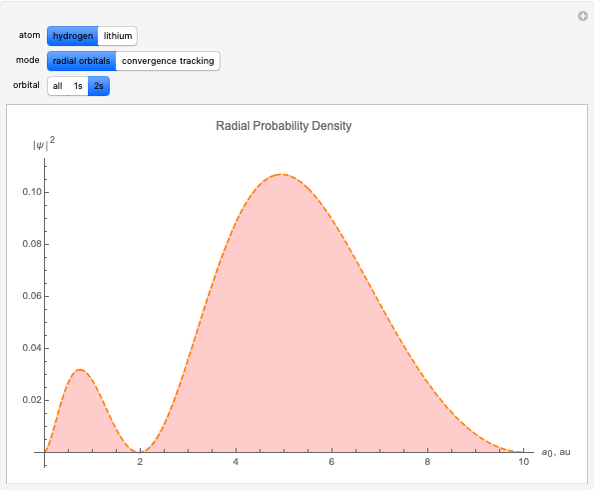
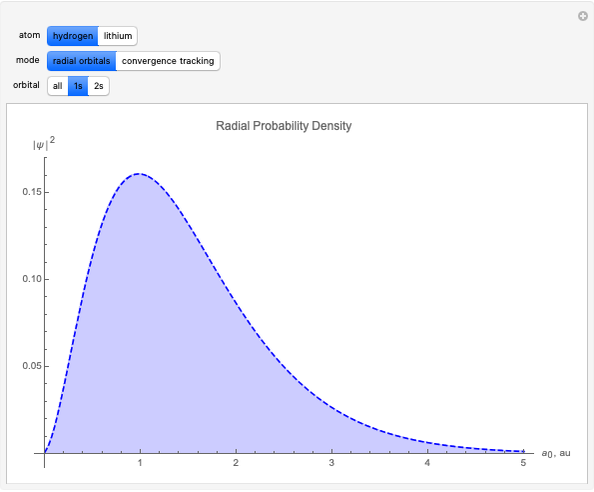
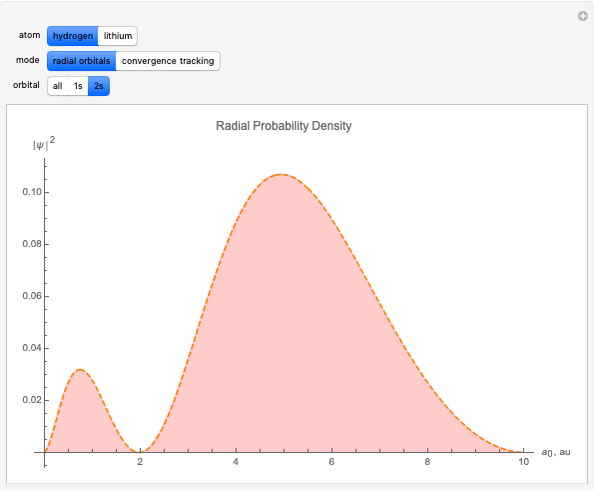
With "radial orbitals" selected, you can choose to view the calculated normalized radial orbital probability distribution functions for hydrogen or lithium atoms.
With orbital "1s" or "2s" selected, you can also view the calculated numerical value for the associated energies by mousing over the curve.
Reference
[1] C. J. Foot, Atomic Physics, New York: Oxford University Press, 2005.
Permanent Citation
 Low-Temperature Heat Capacity of Hydrogen Molecules
Low-Temperature Heat Capacity of Hydrogen Molecules
S. M. Blinder Generalized Unsöld Theorem for Hydrogenic Functions
Generalized Unsöld Theorem for Hydrogenic Functions
S. M. Blinder Hydrogen Orbitals
Hydrogen Orbitals
Michael Trott Schrödinger Equation for a Dirac Bubble Potential
Schrödinger Equation for a Dirac Bubble Potential
S. M. Blinder Unsöld's Theorem
Unsöld's Theorem
S. M. Blinder Rotation-Vibration Energy Level Transitions of a Diatomic Rotor
Rotation-Vibration Energy Level Transitions of a Diatomic Rotor
Whitney R. Hess and Lisa M. Goss (Idaho State University) Rotation-Vibration Transitions of a Parallel Band of a Symmetric Rotor
Rotation-Vibration Transitions of a Parallel Band of a Symmetric Rotor
Whitney R. Hess and Lisa M. Goss Molecular Orbitals for First- and Second-Row Diatomic Molecules
Molecular Orbitals for First- and Second-Row Diatomic Molecules
S. M. Blinder d-Orbitals in an Octahedral Field
d-Orbitals in an Octahedral Field
S. M. Blinder Valence-Bond Theory of the Hydrogen Molecule
Valence-Bond Theory of the Hydrogen Molecule
S. M. Blinder