Simplified Hartree-Fock Computations on Second-Row Atoms



Requires a Wolfram Notebook System
Interact on desktop, mobile and cloud with the free Wolfram Player or other Wolfram Language products.
Modern computational quantum chemistry has developed largely from applications of the Hartree–Fock method to atoms and molecules [1–3]. A simple representation of a many-electron atom is given by a Slater determinant constructed from  occupied spin-orbitals:
occupied spin-orbitals:



















Contributed by: S. M. Blinder (October 2017)
Open content licensed under CC BY-NC-SA
Snapshots
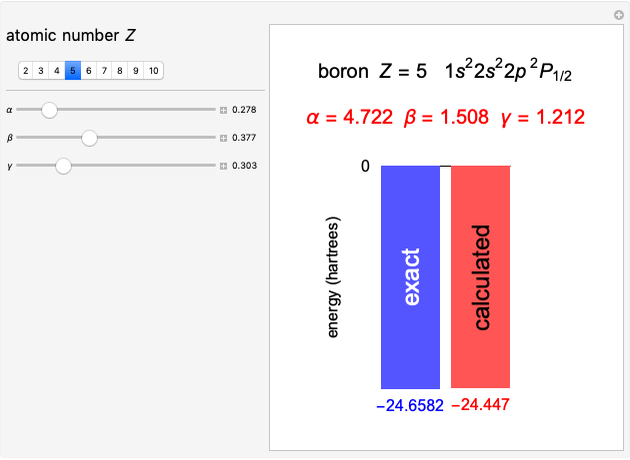
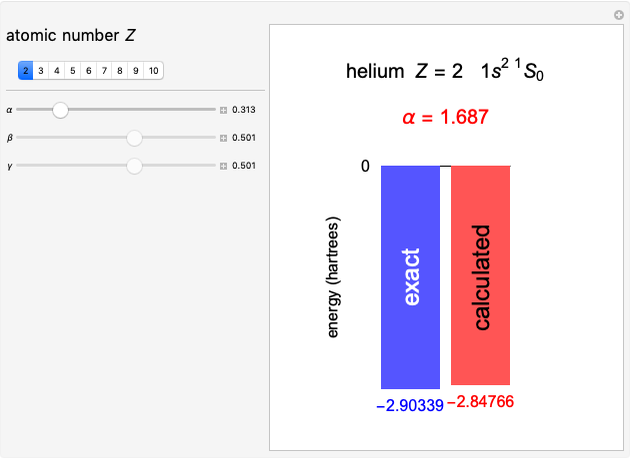
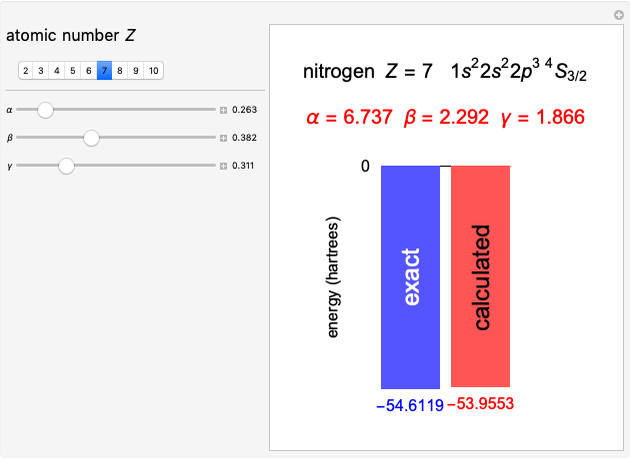
Details
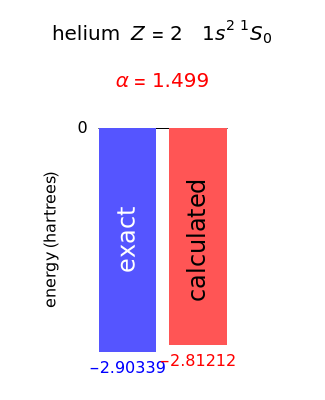
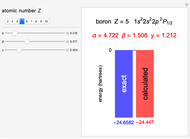
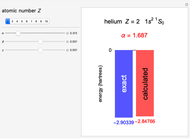
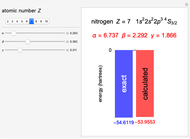
Following are results for optimized functions  ,
,  and
and  . For comparison we also include results from the best Hartree-Fock computations and the exact atomic ground-state energies.
. For comparison we also include results from the best Hartree-Fock computations and the exact atomic ground-state energies.

References
[1] S. M. Blinder, "Simplified Hartree-Fock Computations on Second-Row Atoms," https://arxiv.org/abs/2105.07018
[2] A. Szabo and N. S. Ostlund, Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory, Mineola, NY: Dover, 1996.
[3] S. M. Blinder, "Introduction to the Hartree–Fock Method," in Mathematical Physics in Theoretical Chemistry (S. M. Blinder and J. E. House, eds.), Elsevier, 2018.
Permanent Citation
 Density Functional Computations on Noble Gas Atoms
Density Functional Computations on Noble Gas Atoms
S. M. Blinder Structure and Bonding of Second-Row Hydrides
Structure and Bonding of Second-Row Hydrides
S. M. Blinder Molecular Orbitals for First- and Second-Row Diatomic Molecules
Molecular Orbitals for First- and Second-Row Diatomic Molecules
S. M. Blinder Hydrogen and Lithium Orbitals Using a Hartree Eigenvalue Method
Hydrogen and Lithium Orbitals Using a Hartree Eigenvalue Method
Nikita M. Kostylev WKB Computations on Morse Potential
WKB Computations on Morse Potential
S. M. Blinder Shell Structure of Noble Gas Atoms
Shell Structure of Noble Gas Atoms
S. M. Blinder Hydrides as Isoelectronic Perturbations of the Neon Atom
Hydrides as Isoelectronic Perturbations of the Neon Atom
S. M. Blinder Connection between Quantum-Mechanical Hydrogen Atom and Harmonic Oscillator
Connection between Quantum-Mechanical Hydrogen Atom and Harmonic Oscillator
S. M. Blinder Visualizing Atomic Orbitals
Visualizing Atomic Orbitals
Guenther Gsaller Energy Levels and Wavefunctions of Two Coupled Atomic Rings
Energy Levels and Wavefunctions of Two Coupled Atomic Rings
Vasil Saroka
-
 Barycenter for Three Masses at the Vertices of a Triangle
Barycenter for Three Masses at the Vertices of a Triangle
S. M. Blinder -
 Quantum Particle in a Regular Polygon by Finite Element Method
Quantum Particle in a Regular Polygon by Finite Element Method
S. M. Blinder -
 Three-Qubit W-States on a Quantum Computer
Three-Qubit W-States on a Quantum Computer
S. M. Blinder -
 Boson and Fermion Effects in Helium-Helium Scattering
Boson and Fermion Effects in Helium-Helium Scattering
S. M. Blinder -
 Single-Qubit Quantum Error Correction
Single-Qubit Quantum Error Correction
S. M. Blinder -
 Time Around the World
Time Around the World
S. M. Blinder -
 Kronig-Penney Model with Mathieu Functions
Kronig-Penney Model with Mathieu Functions
S. M. Blinder -
 Kronig-Penney Model with Dirac Comb
Kronig-Penney Model with Dirac Comb
S. M. Blinder -
 Torque Using Cross Product
Torque Using Cross Product
S. M. Blinder -
 Ruler and Compass Construction of a Square with Doubled Area
Ruler and Compass Construction of a Square with Doubled Area
S. M. Blinder -
 Shortest Distance between Two Skew Lines
Shortest Distance between Two Skew Lines
S. M. Blinder -
 Quasi-exact Solution for a Double-Well Potential
Quasi-exact Solution for a Double-Well Potential
S. M. Blinder -
Structure and Bonding of Second-Row Hydrides
S. M. Blinder -
 Unsöld's Theorem
Unsöld's Theorem
S. M. Blinder -
 Polarizability of Hydrogen Atom
Polarizability of Hydrogen Atom
S. M. Blinder -
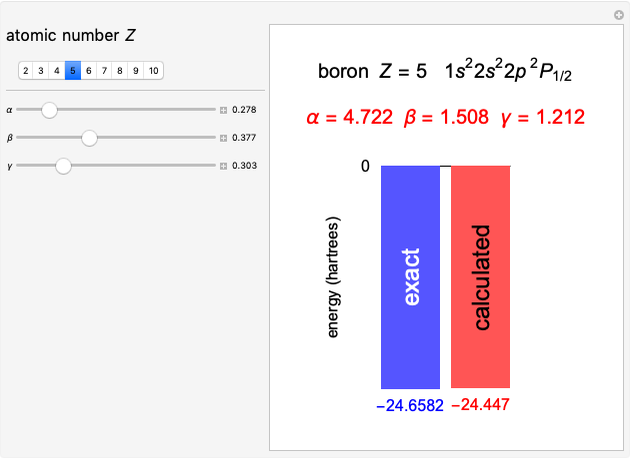 Simplified Hartree-Fock Computations on Second-Row Atoms
Simplified Hartree-Fock Computations on Second-Row Atoms
S. M. Blinder -
 Valence-Bond Theory of the Hydrogen Molecule
Valence-Bond Theory of the Hydrogen Molecule
S. M. Blinder -
 Evolution of Matter from a Quark-Gluon Plasma
Evolution of Matter from a Quark-Gluon Plasma
S. M. Blinder -
 Schrödinger Equation for a Dirac Bubble Potential
Schrödinger Equation for a Dirac Bubble Potential
S. M. Blinder -
 The Higgs Particle
The Higgs Particle
S. M. Blinder