Brillouin Zone of a 2D Square Lattice: Tight Binding Approximation


Requires a Wolfram Notebook System
Interact on desktop, mobile and cloud with the free Wolfram Player or other Wolfram Language products.
Contributed by: Vladimir Gavryushin (Vilnius University, Lithuania) (March 2011)
Open content licensed under CC BY-NC-SA
Snapshots



Details
The energy structure of crystals depends on the interactions between orbitals in the lattice. The tight binding approximation (TB) neglects interactions between atoms separated by large distances, an approximation which greatly simplifies the analysis. In solid-state physics, the TB model calculates the electronic band structure using an approximate set of wave functions based upon superposition of orbitals located at each individual atomic site.
In a TB approximation including only first nearest neighbor s-orbital, the band structure, i.e. the electron energy dispersion in the Brillouin zone of the crystal, is given by
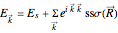 (1)
(1)
where 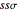 is the overlap integral between s-orbitals,
is the overlap integral between s-orbitals, 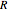 is the translation vector of the lattice, and
is the translation vector of the lattice, and 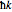 is the "crystal momentum"—the quantum number for periodic systems. The band structure for a simple cubic lattice can now be readily calculated. Assuming that the bond integrals couple only to the first four nearest neighbors with position vectors equal to
is the "crystal momentum"—the quantum number for periodic systems. The band structure for a simple cubic lattice can now be readily calculated. Assuming that the bond integrals couple only to the first four nearest neighbors with position vectors equal to 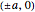 and
and 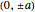 , (1) gives
, (1) gives
 .
.
(a) 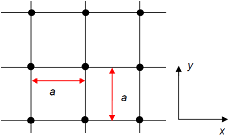
(b) 
(c) 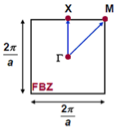
Figure: (a) lattice fragment; (b) four first nearest neighbors included in transfer; (c) the first Brillouin zone.
Setter bars let you compare the lattice electron behavior with the parabolic dispersion curves of the free electrons in free space,
 ,
,
and with the linear dispersion curves of Dirac electrons (as in graphene  -points),
-points),
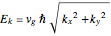 .
.
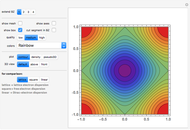
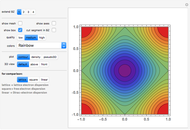
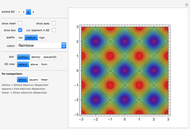
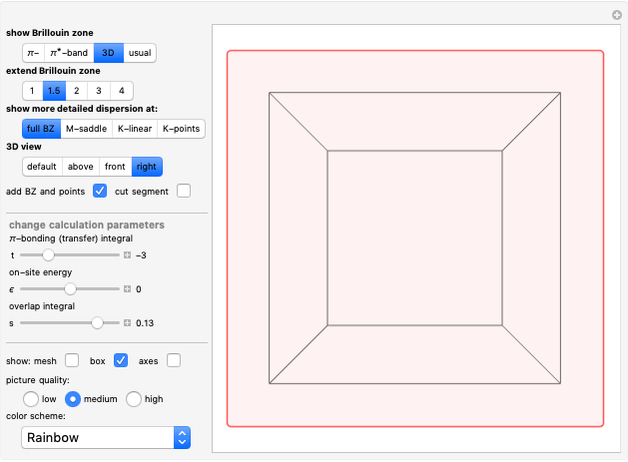
Snapshot 1: constant energy contours for the  -band in the first Brillouin zone of a 2D one-base simple square lattice
-band in the first Brillouin zone of a 2D one-base simple square lattice
Snapshot 2: constant energy contours for the free Dirac electrons in free space
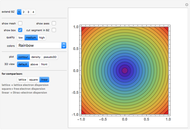
Snapshot 3: constant energy contours for the 2D square lattice in an extended 3×3 Brillouin zone
Snapshot 4: energy dispersion for the -band in an extended 3×3 Brillouin zone of a 2D square lattice
Snapshots 5 and 6: same energy dispersion viewed from top and front
References:
D. G. Pettifor, Bonding and Structure of Molecules and Solids, Oxford: Clarendon Press, 1995.
C. Kittel, Solid State Physics, Hoboken, NJ: John Wiley & Sons, Inc., 1996.
Permanent Citation
 Graphene Brillouin Zone and Electronic Energy Dispersion
Graphene Brillouin Zone and Electronic Energy Dispersion
Vladimir Gavryushin (Vilnius University, Lithuania) X-Ray Reflection by Planes of Atoms in a Cubic Lattice
X-Ray Reflection by Planes of Atoms in a Cubic Lattice
Enrique Zeleny 2D Fourier Transforms
2D Fourier Transforms
John Holland 2D Quantum Problem: Particle in a Disk
2D Quantum Problem: Particle in a Disk
Reinhard Tiebel Exact Numerical Solutions for the Stepped-Infinite-Square Well
Exact Numerical Solutions for the Stepped-Infinite-Square Well
M. Hanson Finite Potential Well
Finite Potential Well
Michael R. Braunstein (Central Washington University)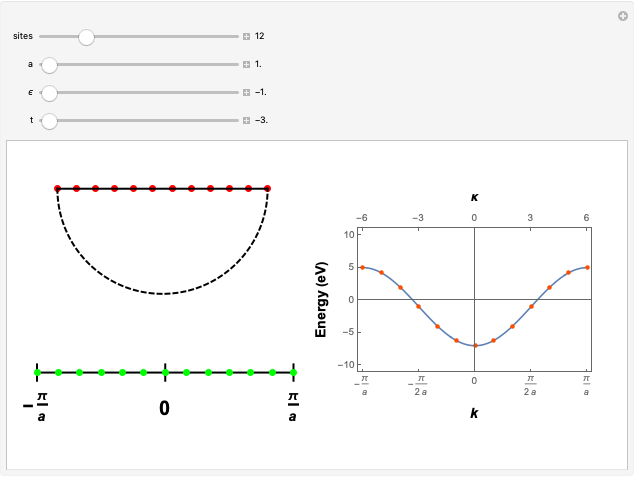 Brillouin Zone Sampling of a Periodic Chain with N Sites
Brillouin Zone Sampling of a Periodic Chain with N Sites
Jessica Alfonsi (University of Padova, Italy) Brillouin Zone Sampling of Chain with N Sites and Modified Periodic Boundary Conditions
Brillouin Zone Sampling of Chain with N Sites and Modified Periodic Boundary Conditions
Jessica Alfonsi (University of Padova, Italy) Wannier Representation for Tight-Binding Hamiltonian of a Periodic Chain with N Sites
Wannier Representation for Tight-Binding Hamiltonian of a Periodic Chain with N Sites
Jessica Alfonsi (University of Padova, Italy) Electronic Structure of 1D and 2D Quasiperiodic Systems
Electronic Structure of 1D and 2D Quasiperiodic Systems
Jessica Alfonsi