Molecular Dynamics of Lennard-Jones Particles Using the Velocity Verlet Algorithm




Requires a Wolfram Notebook System
Interact on desktop, mobile and cloud with the free Wolfram Player or other Wolfram Language products.
Molecular dynamics is a computer simulation technique that follows the time evolution of a set of interacting atoms or molecules by integrating their equations of motion. This Demonstration uses molecular dynamics and the velocity Verlet algorithm to simulate the motion of particles interacting under the Lennard–Jones 6-12 potential. You can vary the temperature, volume, and number of particles to change from the formation of clusters to liquid-like behavior. The color of each particle depends on its speed.
Contributed by: José Luis Gómez-Muñoz (March 2011)
Open content licensed under CC BY-NC-SA
Snapshots
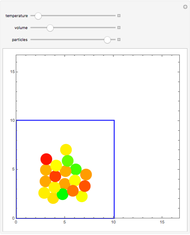
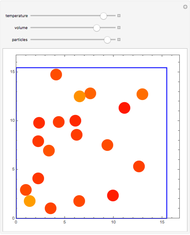
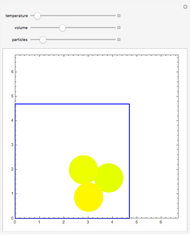
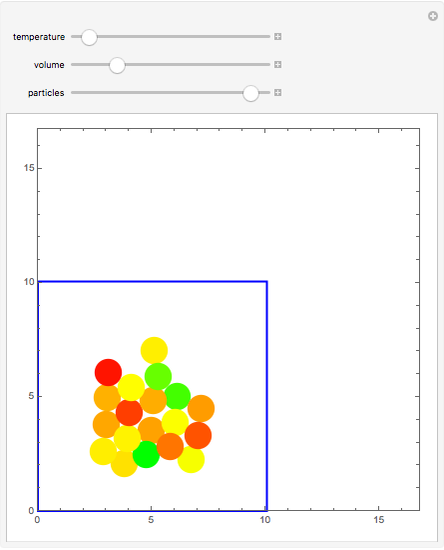
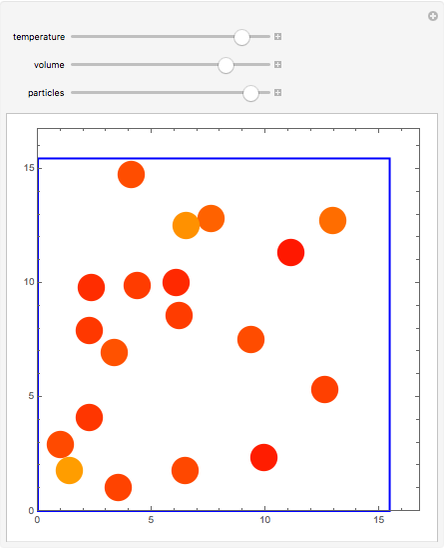
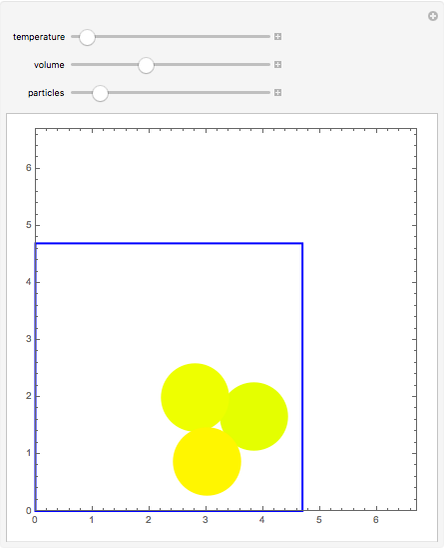
Details
 Diffusion-Limited Aggregation: A Real-Time Agent-Based Simulation
Diffusion-Limited Aggregation: A Real-Time Agent-Based Simulation
Hiroki Sayama Random Walk and Diffusion of Many Independent Particles: An Agent-Based Simulation
Random Walk and Diffusion of Many Independent Particles: An Agent-Based Simulation
Hiroki Sayama Nonlinear Chemical Dynamics: The Peroxidase-Oxidase Reaction
Nonlinear Chemical Dynamics: The Peroxidase-Oxidase Reaction
Corey Knutson-Huddleston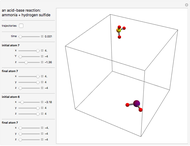 Chemical Reaction Trajectories Using Bézier Curves
Chemical Reaction Trajectories Using Bézier Curves
Mark Mochalsky, Pascual Lahuerta, Jose Vicente Beltran, and Juan Monterde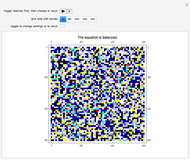 Combustion of Methane Using Block Cellular Automata (BCA) with Feedback
Combustion of Methane Using Block Cellular Automata (BCA) with Feedback
Michael Dewus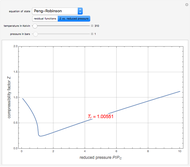 Thermodynamic Properties of Acetylene Using Cubic Equations of State
Thermodynamic Properties of Acetylene Using Cubic Equations of State
Housam Binous and Brian G. Higgins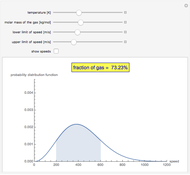 The Maxwell Speed Distribution
The Maxwell Speed Distribution
Jan Fiala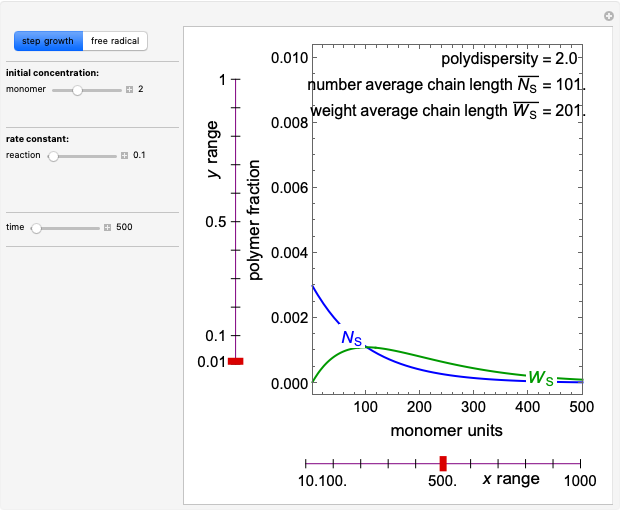 Polymerization in a Batch Reactor
Polymerization in a Batch Reactor
Rachael L. Baumann and Nathan S. Nelson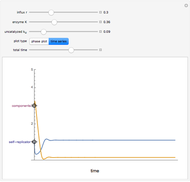 Templator Model of Chemical Self-Replication
Templator Model of Chemical Self-Replication
Enrique Peacock-López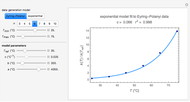 Eyring-Polanyi versus Exponential Model for Chemical Reactions
Eyring-Polanyi versus Exponential Model for Chemical Reactions
Mark D. Normand, Christina S. Barsa, and Micha Peleg
-
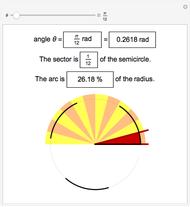 Radians as Percentages
Radians as Percentages
José Luis Gómez-Muñoz -
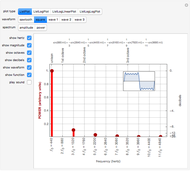 Logarithmic Scales in Acoustic Spectra: Decibels and Octaves
Logarithmic Scales in Acoustic Spectra: Decibels and Octaves
José Luis Gómez-Muñoz -
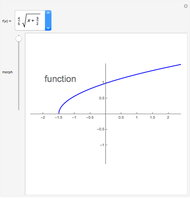 Morphing the Graph of a Function to Its Domain
Morphing the Graph of a Function to Its Domain
José Luis Gómez-Muñoz -
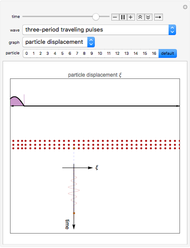 Traveling Longitudinal Pulses and Standing Waves
Traveling Longitudinal Pulses and Standing Waves
José Luis Gómez-Muñoz -
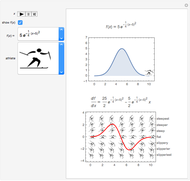 Differentiation for Athletes
Differentiation for Athletes
José Luis Gómez-Muñoz -
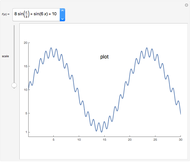 Morphing from Plot to Log Plot
Morphing from Plot to Log Plot
José Luis Gómez-Muñoz -
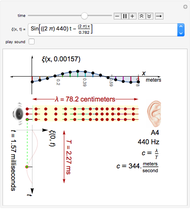 Properties of Acoustic Plane Waves
Properties of Acoustic Plane Waves
José Luis Gómez-Muñoz -
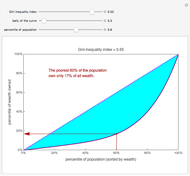 The Lorenz Curve
The Lorenz Curve
José Luis Gómez-Muñoz -
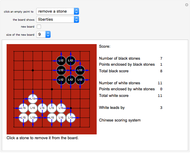 Learn the Game of Go
Learn the Game of Go
José Luis Gómez-Muñoz -
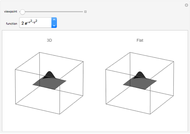 Anamorphic 2D Images That Look Three-Dimensional from a Particular Viewpoint
Anamorphic 2D Images That Look Three-Dimensional from a Particular Viewpoint
José Luis Gómez-Muñoz -
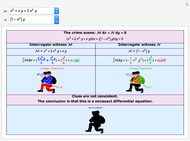 The Murder Mystery Method for Identifying and Solving Exact Differential Equations
The Murder Mystery Method for Identifying and Solving Exact Differential Equations
José Luis Gómez-Muñoz -
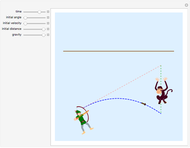 Parabolic Projectile Motion: Shooting a Harmless Tranquilizer Dart at a Falling Monkey
Parabolic Projectile Motion: Shooting a Harmless Tranquilizer Dart at a Falling Monkey
José Luis Gómez-Muñoz -
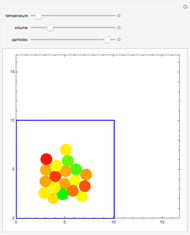 Molecular Dynamics of Lennard-Jones Particles Using the Velocity Verlet Algorithm
Molecular Dynamics of Lennard-Jones Particles Using the Velocity Verlet Algorithm
José Luis Gómez-Muñoz -
 Sailing against the Wind
Sailing against the Wind
José Luis Gómez-Muñoz