Berry Pseudorotation in Phosphorus Pentafluoride




Requires a Wolfram Notebook System
Interact on desktop, mobile and cloud with the free Wolfram Player or other Wolfram Language products.
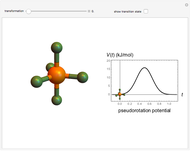
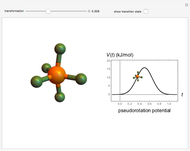
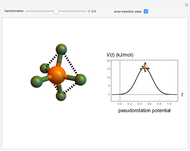
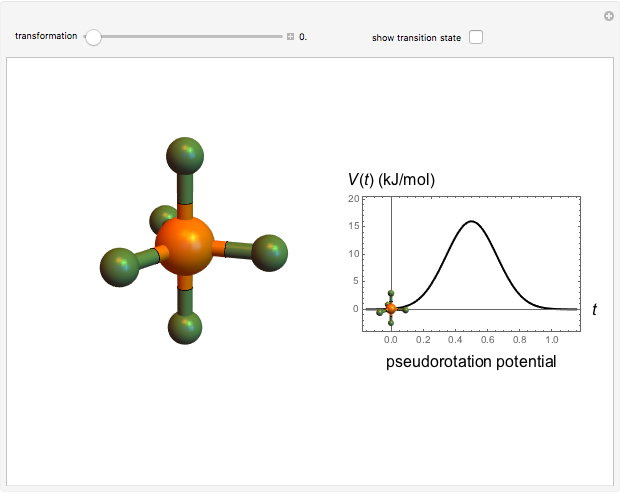
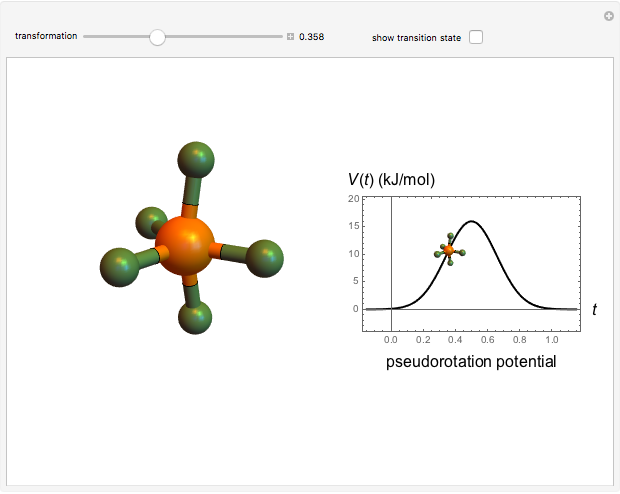
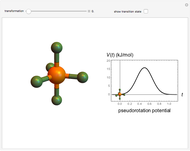
The 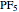 molecule has the configuration of a trigonal bipyramid, as predicted by the VSEPR model. Two of the fluorine atoms, designated as axial, are aligned with the phosphorus atom, with P-F bond distances of 1.58 Å. The remaining three fluorine atoms, designated as equatorial, are arranged in an equilateral triangle with P-F bond distances of 1.53 Å. Even though there exist two geometrically inequivalent types of fluorine atoms, the NMR spectrum of the molecule shows but a single resonance frequency. This indicates that the axial and equatorial
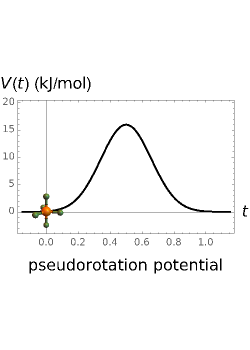
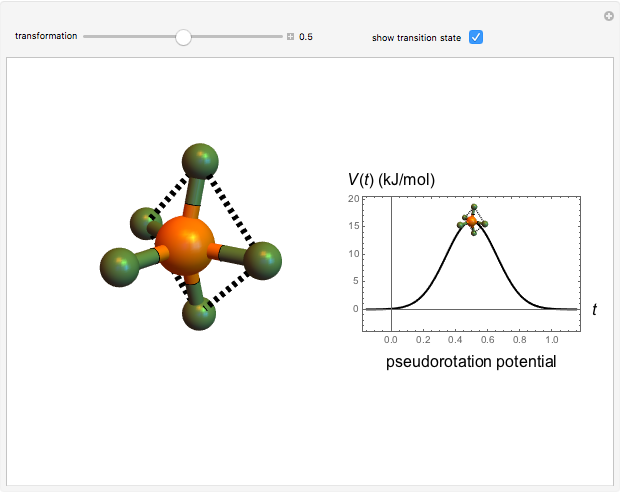
molecule has the configuration of a trigonal bipyramid, as predicted by the VSEPR model. Two of the fluorine atoms, designated as axial, are aligned with the phosphorus atom, with P-F bond distances of 1.58 Å. The remaining three fluorine atoms, designated as equatorial, are arranged in an equilateral triangle with P-F bond distances of 1.53 Å. Even though there exist two geometrically inequivalent types of fluorine atoms, the NMR spectrum of the molecule shows but a single resonance frequency. This indicates that the axial and equatorial  nuclei exchange their environments more rapidly than the time it takes to make the NMR measurement. Thus only a single resonance peak is seen. R. S. Berry [1] proposed a mechanism for atomic rearrangement, known as pseudorotation, since it simulates a rotation of the axial direction of the molecule. According to the proposed mechanism, one of the equatorial bonds serves as a pivot while the two axial bonds bend into two sides of an equilateral triangle. Simultaneously, the remaining two equatorial bonds line up to become the new axial bonds. Quantum-chemical computations predict a barrier of approximately 16 kJ/mol [2], which permits rapid tunneling between configurations at room temperature.
nuclei exchange their environments more rapidly than the time it takes to make the NMR measurement. Thus only a single resonance peak is seen. R. S. Berry [1] proposed a mechanism for atomic rearrangement, known as pseudorotation, since it simulates a rotation of the axial direction of the molecule. According to the proposed mechanism, one of the equatorial bonds serves as a pivot while the two axial bonds bend into two sides of an equilateral triangle. Simultaneously, the remaining two equatorial bonds line up to become the new axial bonds. Quantum-chemical computations predict a barrier of approximately 16 kJ/mol [2], which permits rapid tunneling between configurations at room temperature.
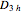
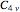
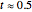
Contributed by: S. M. Blinder (August 2018)
Open content licensed under CC BY-NC-SA
Details
References
[1] R. S. Berry, "Correlation of Rates of Intramolecular Tunneling Processes, with Application to Some Group V Compounds," The Journal of Chemical Physics, 32(3), 1960 pp. 933–938. doi:10.1063/1.1730820.
[2] C. J. Marsden, "Pseudorotation Pathway and Quadratic Force Field for 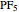 , by ab initio Calculations," Journal of the Chemical Society, Chemical Communications, 1984(7), pp. 401–402. doi:10.1039/C39840000401.
, by ab initio Calculations," Journal of the Chemical Society, Chemical Communications, 1984(7), pp. 401–402. doi:10.1039/C39840000401.
Snapshots
Permanent Citation
 Absorption Spectroscopy
Absorption Spectroscopy
S. M. Blinder Internal Rotation in Ethane and Substituted Analogs
Internal Rotation in Ethane and Substituted Analogs
S. M. Blinder Statistical Thermodynamics of Ideal Gases
Statistical Thermodynamics of Ideal Gases
S. M. Blinder Bonding and Antibonding Molecular Orbitals
Bonding and Antibonding Molecular Orbitals
S. M. Blinder Configuration Interaction for the Helium Isoelectronic Series
Configuration Interaction for the Helium Isoelectronic Series
S. M. Blinder Structure and Bonding of Second-Row Hydrides
Structure and Bonding of Second-Row Hydrides
S. M. Blinder Simplified Hartree-Fock Computations on Second-Row Atoms
Simplified Hartree-Fock Computations on Second-Row Atoms
S. M. Blinder Molarity of Aqueous Salt Solutions
Molarity of Aqueous Salt Solutions
S. M. Blinder Ions with Noble Gas Configurations
Ions with Noble Gas Configurations
S. M. Blinder Three-Parameter Variational Wavefunctions for the Helium Isoelectronic Series
Three-Parameter Variational Wavefunctions for the Helium Isoelectronic Series
S. M. Blinder
-
 Mona Lisa and the Golden Rectangle
Mona Lisa and the Golden Rectangle
S. M. Blinder -
 Principal Major and Minor Scales on a Piano
Principal Major and Minor Scales on a Piano
S. M. Blinder -
 Generating Near-Isosceles Pythagorean Triples
Generating Near-Isosceles Pythagorean Triples
S. M. Blinder -
 Landau Levels in a Magnetic Field
Landau Levels in a Magnetic Field
S. M. Blinder -
 The Wilberforce Pendulum
The Wilberforce Pendulum
S. M. Blinder -
 Eigenstates for a Hydrogen Atom Confined to an Infinite Spherical Potential Well
Eigenstates for a Hydrogen Atom Confined to an Infinite Spherical Potential Well
S. M. Blinder -
 Bohr's Model for the Hydrogen Molecule
Bohr's Model for the Hydrogen Molecule
S. M. Blinder -
 Analogies between Kinematics of Linear and Angular Motion
Analogies between Kinematics of Linear and Angular Motion
S. M. Blinder -
 Orbital Transformations in Diels-Alder Reaction
Orbital Transformations in Diels-Alder Reaction
S. M. Blinder -
 Collision of Two Droplets
Collision of Two Droplets
S. M. Blinder -
 Alternative Forms of Maxwell's Equations
Alternative Forms of Maxwell's Equations
S. M. Blinder -
 A New Type of Polyhedron: The Scutoid
A New Type of Polyhedron: The Scutoid
S. M. Blinder -
 Uranium Enrichment Using Gas Centrifuges
Uranium Enrichment Using Gas Centrifuges
S. M. Blinder -
 Asymptotic Expansions for Some Special Functions
Asymptotic Expansions for Some Special Functions
S. M. Blinder -
 Berry Pseudorotation in Phosphorus Pentafluoride
Berry Pseudorotation in Phosphorus Pentafluoride
S. M. Blinder -
 The Multiplication Parabola
The Multiplication Parabola
S. M. Blinder -
 Quantum Pendulum
Quantum Pendulum
S. M. Blinder -
 Generalized Fibonacci Sequence and the Golden Ratio
Generalized Fibonacci Sequence and the Golden Ratio
S. M. Blinder -
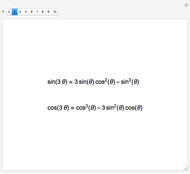 Multiple Angle Formulas for Sine and Cosine
Multiple Angle Formulas for Sine and Cosine
S. M. Blinder -
 Rule of Product Applied to Decks of Cards
Rule of Product Applied to Decks of Cards
S. M. Blinder