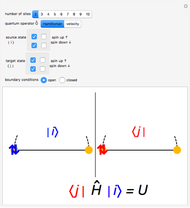
Hubbard Model Interactive Calculator for 1D Systems




Requires a Wolfram Notebook System
Interact on desktop, mobile and cloud with the free Wolfram Player or other Wolfram Language products.
This Demonstration shows the basics of the fermion Hubbard model applied to one-dimensional systems such as chains with  sites with either open or closed boundary conditions and any phase
sites with either open or closed boundary conditions and any phase  at the ends. The Hubbard model provides a very simple but physically powerful description of electronic many-body effects in quantum mechanical systems. This can be understood by considering the form of its Hamiltonian, expressed in second quantization formalism:
at the ends. The Hubbard model provides a very simple but physically powerful description of electronic many-body effects in quantum mechanical systems. This can be understood by considering the form of its Hamiltonian, expressed in second quantization formalism:  , where
, where  and
and  are site indices,
are site indices,  are all pairs of first nearest neighbors sites,
are all pairs of first nearest neighbors sites,  gives the number of electrons on site with spin
gives the number of electrons on site with spin  ,
,  and
and  are electron creation and annihilation operators, respectively, and
are electron creation and annihilation operators, respectively, and  and
and  are positive interaction constants, respectively. The term is a single-particle (tight-binding) term (thus it contains no many-body features) and describes the hopping of electrons localized on atomic-like orbitals between nearest neighbor sites and models the kinetic energy of the system. The
are positive interaction constants, respectively. The term is a single-particle (tight-binding) term (thus it contains no many-body features) and describes the hopping of electrons localized on atomic-like orbitals between nearest neighbor sites and models the kinetic energy of the system. The  term gives a potential energy contribution to the Hamiltonian and models the Coulomb repulsion between two electrons with opposite spin in the same orbital, hence it is the effective many-body (two-body) term. Another related operator is the velocity operator
term gives a potential energy contribution to the Hamiltonian and models the Coulomb repulsion between two electrons with opposite spin in the same orbital, hence it is the effective many-body (two-body) term. Another related operator is the velocity operator  , where
, where  is the lattice parameter of the chain and
is the lattice parameter of the chain and  is the Planck constant (since these are normalization constants, they have been set equal to 1 in the program). The term
is the Planck constant (since these are normalization constants, they have been set equal to 1 in the program). The term  represents the current operator, which is relevant for investigating optical conductivity properties.
represents the current operator, which is relevant for investigating optical conductivity properties.








Contributed by: Jessica Alfonsi (University of Padova, Italy) (March 2011)
Open content licensed under CC BY-NC-SA
Snapshots



Details
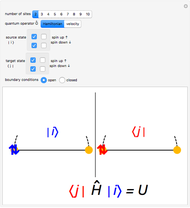
Snapshot 1: Hamiltonian matrix element between two equal states with one doubly occupied site
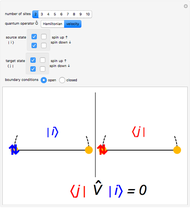
Snapshot 2: velocity matrix element between a doubly occupied state and a state with one electron on each site
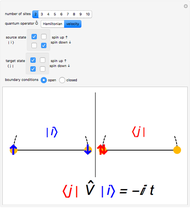
Snapshot 3: velocity matrix element between two equal states with one doubly occupied site
Part of the initialization is taken from the following book: W. Kinzel and G. Reents, Physics by Computer: Programming Physical Problems Using Mathematica and C, New York: Springer, 1998.
H. Q. Lin and J. E. Gubernatis, "Exact Diagonalization Method for Quantum Systems," Computers in Physics 7(4), 1993 p. 400.
J. Alfonsi, "Small Crystal Models for the Electronic Properties of Carbon Nanotubes," Ph.D. thesis, University of Padova, 2009, Chapter 6 and references therein.
Permanent Citation
 Models for Edge States in the Electronic Spectra of Dimer Chains
Models for Edge States in the Electronic Spectra of Dimer Chains
Jessica Alfonsi Quantized Solutions of the 1D Schrödinger Equation for a Harmonic Oscillator
Quantized Solutions of the 1D Schrödinger Equation for a Harmonic Oscillator
Jamie Williams Worm Algorithm for J-Current Model
Worm Algorithm for J-Current Model
Darya Aleinikava Retrocausality: A Toy Model
Retrocausality: A Toy Model
Peter Evans Energy Levels of a Quantum Harmonic Oscillator in Second Quantization Formalism
Energy Levels of a Quantum Harmonic Oscillator in Second Quantization Formalism
Jessica Alfonsi (University of Padova, Italy) Brillouin Zone of a 2D Square Lattice: Tight Binding Approximation
Brillouin Zone of a 2D Square Lattice: Tight Binding Approximation
Vladimir Gavryushin (Vilnius University, Lithuania) Normal Modes in a Periodic Monatomic Linear Chain
Normal Modes in a Periodic Monatomic Linear Chain
Luis Elcoro (University of the Basque Country, Spain) Finite Potential Well
Finite Potential Well
Michael R. Braunstein (Central Washington University) Rotation-Vibration Transitions for a Perpendicular Band of a Symmetric Rotor
Rotation-Vibration Transitions for a Perpendicular Band of a Symmetric Rotor
Whitney R. Hess and Lisa M. Goss Rotation-Vibration Transitions of a Parallel Band of a Symmetric Rotor
Rotation-Vibration Transitions of a Parallel Band of a Symmetric Rotor
Whitney R. Hess and Lisa M. Goss
-
 Evolution in a Cellular-Automaton Model of Gray-Scott Reaction-Diffusion System
Evolution in a Cellular-Automaton Model of Gray-Scott Reaction-Diffusion System
Jessica Alfonsi -
 Kuramoto Model for Phase Locking of Coupled Oscillators
Kuramoto Model for Phase Locking of Coupled Oscillators
Jessica Alfonsi -
 Topological Winding Number in 1D Su-Schrieffer-Heeger Model
Topological Winding Number in 1D Su-Schrieffer-Heeger Model
Jessica Alfonsi -
Models for Edge States in the Electronic Spectra of Dimer Chains
Jessica Alfonsi -
 Moiré Patterns and Commensurability in Rotated Graphene Bilayers
Moiré Patterns and Commensurability in Rotated Graphene Bilayers
Jessica Alfonsi -
 Electronic Structure of 1D and 2D Quasiperiodic Systems
Electronic Structure of 1D and 2D Quasiperiodic Systems
Jessica Alfonsi -
 Labyrinth Tiling from Quasiperiodic Octonacci Chains
Labyrinth Tiling from Quasiperiodic Octonacci Chains
Jessica Alfonsi -
 Self-Similar Qubistic Plot of the S_z=0 Half-Filled Hubbard Model Basis States
Self-Similar Qubistic Plot of the S_z=0 Half-Filled Hubbard Model Basis States
Jessica Alfonsi -
 Electronic Band Structure of Armchair and Zigzag Graphene Nanoribbons
Electronic Band Structure of Armchair and Zigzag Graphene Nanoribbons
Jessica Alfonsi -
 Electronic Structure of a Single-Walled Carbon Nanotube in Tight-Binding Wannier Representation
Electronic Structure of a Single-Walled Carbon Nanotube in Tight-Binding Wannier Representation
Jessica Alfonsi -
 Metallicity Condition and Electronic Density of States in Achiral Single-Walled Carbon Nanotubes
Metallicity Condition and Electronic Density of States in Achiral Single-Walled Carbon Nanotubes
Jessica Alfonsi -
 Rate Equations for a Three-Level Model in Low-Dimensional Exciton Systems
Rate Equations for a Three-Level Model in Low-Dimensional Exciton Systems
Jessica Alfonsi -
 Hubbard Model Interactive Calculator for 1D Systems
Hubbard Model Interactive Calculator for 1D Systems
Jessica Alfonsi -
 Electronic Band Structure of a Single-Walled Carbon Nanotube by the Zone-Folding Method
Electronic Band Structure of a Single-Walled Carbon Nanotube by the Zone-Folding Method
Jessica Alfonsi -
 Optical Matrix Elements of Single-Walled Carbon Nanotubes for Longitudinal Polarization of Light
Optical Matrix Elements of Single-Walled Carbon Nanotubes for Longitudinal Polarization of Light
Jessica Alfonsi -
 Optical Properties of Graphene
Optical Properties of Graphene
Jessica Alfonsi -
 Finite Element Solution of 1D Poisson Equation with Galerkin Spectral Decomposition
Finite Element Solution of 1D Poisson Equation with Galerkin Spectral Decomposition
Jessica Alfonsi -
Energy Levels of a Quantum Harmonic Oscillator in Second Quantization Formalism
Jessica Alfonsi -
 Brillouin Zone Sampling of Chain with N Sites and Modified Periodic Boundary Conditions
Brillouin Zone Sampling of Chain with N Sites and Modified Periodic Boundary Conditions
Jessica Alfonsi -
 Brillouin Zone of a Single-Walled Carbon Nanotube
Brillouin Zone of a Single-Walled Carbon Nanotube
Jessica Alfonsi